NBCR – National Biomedical Computation Resource was founded in 1994 to facilitate interaction among biomedical scientists and promote the development of computing power at the national supercomputer centers. Since that time, NBCR has led the development of new technology in computing to benefit the biomedical research community and bridge the gap between emerging information technologies and NIH-funded science across diverse biomedical research areas.
NBCR’s vision centers on the development of tools and technologies that enable models to bridge across diverse scales of biological organization that ranges from molecules to organs systems to gain a quantitative understanding of biological function and phenotypes, while enabling biomedical science to take full advantage of all types and sources of relevant data. Our products raise from our Driving Biomedical Projects, and is applied to a board scientific community.
Click here for the list of NBCR Software, WebServices, Workflows, and Software Rolls
With the click of a button, a simulation is triggered and a user can visualize the dance a protein performs in a solution. Described above is the combined power of Kepler and Molecular Dynamic (MD) simulations. MD simulation is a powerful tool to study the atomistic details of a biological system. Atom movements across time are…
CRISPR-Cas9 conformational activation, as revealed from Gaussian accelerated Molecular Dynamics (GaMD) simulations. GaMD reveals the conformational change of the HNH domain (green) from its inactive configuration (top) to the active state (bottom). This conformational change, which occurs along milliseconds, is captured with atomic-level precision via the use of advanced simulation methods (i.e. GaMD) and state-of-the-art…
There’s long been a standard tradeoff in biochemistry: You can study overall shape (of, say, a macromolecule or an organelle) or high-resolution detail within, say, at the 10-40-nanometer scale. Seeing both at the same time would be one of today’s scientific Holy Grails. But new work by a research team at the National Biomedical Computation…
The ability to locate and visualize proteins and macromolecular complexes in cells and tissues in 3D high resolution continues to be a challenge in biomedical studies. Various techniques and tools are key to this work. For example, light microscopy uses fluorescent labels to track elements of interest, but it provides an incomplete view, including only…
Computational models of virions were generated using cellPACK. From left to right, the five models illustrate a random distribution of an increasing number of spikes: 10 (average number of spikes for HIV-1 virions), 49 (average number of spikes for V1 hVLPs), 127 (average number of spikes for V4 hVLPs), 214 (highest number of spikes estimated…
Reprint of article originally published January 3, 2017 in c&en By Katherine Bourzac Determining the structures of enzymes and other biomolecules with X-ray crystallography has deepened biologists’ understanding of the inner workings of cells and led to the design of many important drugs. Increasingly, researchers are using computer modeling to attain a more realistic picture of…
Reprint of article originally published December 9, 2016 from The University of Chicago By Robert Mitchum Despite persistent unemployment in the United States, millions of jobs are hard to fill due to a lack of qualified applicants. While community college and training organizations seek to equip people with the skills required for these openings, it’s a…
Reprint of article originally published November 11, 2016 from The Scripps Research Institute Three groups at The Scripps Research Institute (TSRI) have been awarded grants from the National Institutes of Health (NIH) to develop methods for computational modeling and to apply them to cutting-edge systems in biology and health. “The three projects are highly symbiotic,…
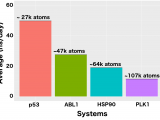
Reprint of article originally published October 27, 2016 from the Texas Advanced Computing Center By Faith Singer-Villalobos Even though it’s almost impossible to see, computational biophysicist Rommie Amaro is using the Stampede supercomputer at the Texas Advanced Computing Center at The University of Texas at Austin to model the largest atomic level system of the tumor suppression protein p53 to…
Reprint of article originally published October 5, 2016 in AAAS By Robert F. Service It has been nearly impossible to get a good look at Rommie Amaro’s favorite protein in action. Called p53, the protein sounds the alarm to kill cells with DNA damage and prevent them from becoming cancerous—one reason why it has been called…